Bioinformatische Verfahren zur kombinierten Analyse von Transkriptom-, Metabolom- und Proteomdaten
Forschungsbericht (importiert) 2003 - Max-Planck-Institut für Molekulare Pflanzenphysiologie
Kombinierte und integrative Datenanalyse
In der modernen Biologie spielt die Bioinformatik eine immer bedeutendere Rolle [1]. Aus dem Bereich Pflanzenforschung entstehen neue Herausforderungen an dieses junge Wissenschaftsgebiet durch die Notwendigkeit, Daten integrativ zu analysieren, die von Genom-, Transkriptom-, Proteom- und Metabolom-Technologien in Form von Profilen erzeugt werden und die eine direkte Verbindung zwischen traditioneller Genetik und beobachteten Phänotypen von Pflanzen ermöglichen [2].
In einer solchen integrativen Analyse untersuchten Joachim Selbig und sein Team Metabolit-Profile von Zellen aus Kartoffelknollen, die aus Gaschromatograph-Massenspektren gewonnen wurden, und Genexpressions-Profile, die mit Nylonfilter-Arrays bestimmt wurden. Verschiedene Entwicklungsstadien dieses biologischen Systems konnten durch Metabolit-Profile mit einer höheren Genauigkeit unterschieden werden als durch Genexpressions-Profile [3].
Die Erfahrungen, die die Wissenschaftler bei dieser Untersuchung gewonnen haben, fanden bei der Entwicklung des Softwaresystems MetaGeneAlyse Berücksichtigung [4]. MetaGeneAlyse ist ein Server-basiertes System, das über ein Webinterface zugänglich ist (vergleiche Abb. 1). Das System bietet einerseits eine Vielzahl von Optionen zur Normalisierung, Dimensionsreduktion, Clusterung und Visualisierung von Daten, andererseits garantiert es eine leichte Bedienbarkeit. Zu den zur Verfügung stehenden Clustermethoden gehören K-Means- und hierarchische Clusterverfahren mit verschiedenen Abstandsmaßen zur Gruppierung der Datenpunkte. Neben dem Euklidischen Abstand und dem Korrelationskoeffizienten kann auch die wechselseitige Information (mutual information) als Abstandsmaß ausgewählt werden.
.")
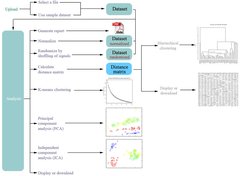
Die wechselseitige Information ist ein allgemeines Entropie-basiertes Maß zur Beschreibung der Abhängigkeit von Variablen. Sie ist insbesondere geeignet, nicht-lineare Beziehungen auszudrücken. Die Wissenschaftler erweiterten den Histogramm-basierten Ansatz zur Verarbeitung kontinuierlicher Daten, indem sie eine Unschärfe bei der Diskretisierung der kontinuierlichen Messwerte einführten [5].
Angewandt wurde das Verfahren auf öffentlich zugängliche Hefe-Genexpressions-Daten und auf Daten zur Untersuchung von Schwefelmangel in Arabidopsis thaliana. Ein auf der Grundlage der Clusterung von Genexpressions-Profilen erzeugtes Netzwerk spiegelt physiologische Aspekte der Stressantwort in biologisch sinnvoller Weise wider.
Die wechselseitige Information wurde auch als Maß verwendet, um die Organismen-spezifische Konservierung von Aminosäuren in funktionalen Motiven von Proteinsequenzen zu bewerten [6]. Am Beispiel von Kinasen zeigten die Wissenschaftler Zusammenhänge zwischen sequentieller und struktureller Konserviertheit auf.
Normalisierung
Die integrative Analyse von Daten, die durch verschiedene Technologien erzeugt wurden, erfordert eine Normalisierung der Daten vor der eigentlichen Verarbeitung, zum Beispiel durch Clusterverfahren. Liegen die Daten in Form von Quotienten aus Messwerten über ein Behandlungsexperiment und einen Wildtyp vor, wird häufig eine Normalisierung durch Logarithmusbildung erreicht. Andere Normalisierungen basieren auf der Vektornorm, der Rangordnung, der einheitlichen Varianz oder dem Z-Score. In dem bereits erwähnten Softwaresystem MetaGeneAlyse werden verschiedene Datennormalisierungen zur Verfügung gestellt.
In einer Untersuchung zur paarweisen Unterscheidung der Ausgangslinien (Parentallinien) und der segregierenden Nachkommen (Filialgeneration) bei einem Arabidopsis-Kreuzungs-Experiment auf der Grundlage von Metabolit-Profilen erwies sich die Vektornorm als die am besten diskriminierende [7]. Dieser Normalisierung entspricht eine Projektion auf eine Hyperkugel und kompensiert daher Intensitätsunterschiede; sie steht in Beziehung zur häufig angewandten Korrelationsanalyse.
Dimensionsreduktion
Durch die große Anzahl von Genen und Metaboliten als Variable entstehen bei der integrativen Analyse von Genexpressions- und Metabolit-Profilen hochdimensionale Merkmalsräume. Im Vergleich zur großen Anzahl von Variablen ist die Anzahl der zur Verfügung stehenden Datenpunkte häufig sehr klein. Die dadurch entstehenden Probleme werden unter dem Ausdruck "Fluch der Dimensionalität" (curse of dimensionality) zusammengefasst. In hochdimensionalen Räumen streuen Datenpunkte beträchtlich, sodass sich Clusterungen nur in Unterräumen niedriger Dimensionalität ergeben. Zur Analyse von Genexpressions- und Metabolit-Profilen werden daher vielfach zunächst Dimensionsreduktionsverfahren eingesetzt.
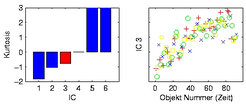
Bei der Analyse der Daten des bereits erwähnten Arabidopsis-Kreuzungs-Experiments wurde eine Kombination aus Hauptkomponentenanalyse (Principal Component Analysis, PCA) und Unabhängiger Komponentenanalyse (Independent Component Analysis, ICA) vorgeschlagen [7]. Für die ersten beiden berechneten unabhängigen Komponenten ergab sich eine biologisch sinnvolle Interpretation. Darüber hinaus zeigt Abbildung 2, dass die dritte unabhängige Komponente (IC3) vom Zeitpunkt der Probenverarbeitung im Massenspektrometer abhängt und daher ein Hinweis auf eine zunehmende Verunreinigung des Geräts sein könnte. Zur Bewertung der unabhängigen Komponenten wurde die Kurtosis als statistisches Maß verwendet, das die Abweichung einer Verteilung von einer Normalverteilung ausdrückt.
 bei einem Arabidopsis-Kreuzungs-Experiment.")
Visualisierung
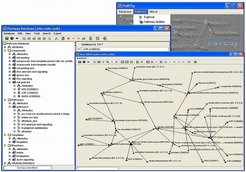
Ein Ziel der Analyse von Profildaten besteht in der Konstruktion und Rekonstruktion von metabolischen und regulatorischen Netzwerken. Durch Clusterung von Genexpressions- und Metabolit-Profilen sollen einerseits Hypothesen über neue Pfade beziehungsweise Netzwerke erzeugt werden, andererseits soll bekanntes Wissen über metabolische und regulatorische Zusammenhänge genutzt werden, um neue Funktionszuordnungen auf der Grundlage solcher Clusterungen zu überprüfen. Diese Zusammenhänge liegen in Form von Datenbanken über metabolische und regulatorische Netzwerke vor, die teilweise fehlerhafte oder unvollständige Informationen enthalten. Mit dem Softwaresystem PaVESy (Pathway Visualization and Editing System) haben Joachim Selbig und sein Team eine Möglichkeit geschaffen, Pfade und Netzwerke zu editieren und zu visualisieren [8]. PaVESy basiert auf einer relationalen Datenbank, die Informationen über Gene, Proteine, Metabolite und chemische Reaktionen enthält. Abbildung 3 zeigt exemplarisch das Ergebnis der Suche nach einer Reaktionsumgebung der Substanz Pyrophosphat in der Reaktionsdatenbank.
.")
Die bereits angesprochene große Anzahl von Variablen bei der integrativen Analyse von Genexpressions- und Metabolit-Profilen erschwert die Interpretation der Analyseergebnisse, zum Beispiel beim Vergleich der Reaktion einer Pflanze auf eine Behandlung gegenüber einem Wildtyps. Möglichkeiten zur Unterstützung dieser Interpretationen bieten geeignete Visualisierungen. Das am MPI für molekulare Pflanzenphysiologie entwickelte Softwaresystem MapMan bildet große Mengen von Daten über Gen-Expressionen und Metabolit-Konzentrationen auf schematische Pfade oder Netzwerke ab [9]. Abbildung 4 illustriert das System.
.")
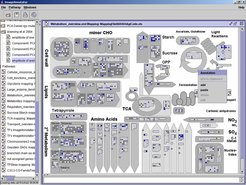
Das Softwaresystem besteht aus verschiedenen Modulen zur Strukturierung von Daten in funktionelle Kategorien und zu ihrer Visualisierung gemäß der Kategorisierung. Die Funktionszuordnung erfolgt auf der Grundlage verfügbarer Annotationen und manueller Korrektur.
Integrative Analyse von Daten, die von Genom-, Transkriptom-, Proteom- und Metabolom-Technologien erzeugt werden, bedeutet sowohl die Zusammenführung der Daten verschiedenen Typs als auch die Einbeziehung von Daten aus verschiedenen Quellen. Mit der Datenbank CSB.DB (Comprehensive Systems-Biology Database) steht eine Ressource zur Verfügung, die Informationen über das Co-Response-Verhalten von Genen enthält, die durch statistische Analyse einer Vielzahl von öffentlich zugänglichen Datenbanken über Genexpressions-Experimente berechnet wurden. Die in CSB.DB enthaltenen Co-Response-Informationen erlaubten zum Beispiel eine verbesserte Vorhersage der Operonstruktur aus Genexpressions-Daten [10].
Mit der Zunahme der Verfügbarkeit von Daten über Genexpressionen, Metabolitkonzentrationen und Enzymaktivitäten wird es möglich sein, mit überwachten Lernverfahren robuste Marker zu identifizieren, die eine genaue Diagnose des physiologischen Zustands einer Pflanze zulassen. Mit einer entsprechenden diagnostischen Plattform können Umwelteinflüsse schneller erkannt und optimale Wachstums- und Ertragsbedingungen genauer bestimmt werden. Das Team um Joachim Selbig hat bereits erste Schritte unternommen, um ein solches Softwaresystem zu entwickeln.
Literatur
[1] Yu, U., S. H. Lee, Y. J. Kim and S. Kim: Bioinformatics in the Post-genome Era. Journal of Biochemisty and Molecular Biology 37, 75-82 (2004).
[2] Edwards, D. and J. Batley: Plant bioinformatics: from genome to phenome. Trends in Biotechnology 22, 232-237 (2004).
[3] Urbanczyk-Wochniak, E., A. Lüdemann, J. Kopka, J. Selbig, U. Rössner-Tunali, L. Willmitzer and A. R. Fernie: Parallel analysis of transcript and metabolic profiles: a new approach in systems biology. EMBO Reports 4, 989-993 (2003).
[4] Daub, C.O., S. Kloska and J. Selbig: MetaGeneAlyse. Bioinformatics 19, 2332-2333 (2003).
[5] Steuer, R., J. Kurths, C. Daub, J. Weise and J. Selbig: The mutual information: Detecting and evaluating dependencies between variables. Bioinformatics 18, S231-S240 (2002).
[6] Weckwerth, W. and J. Selbig: Scoring and identifying organism-specific functional patterns and putative phosphorylation sites in protein sequences using mutual information. Biochemical and Biophysical Research Communications 307, 516-521 (2003).
[7] Scholz, M., S. Gatzek, A. Sterling, O. Fiehn and J. Selbig: Metabolite fingerprinting: detecting biological features by Idependent Component Analysis. Bioinformatics Advance Access April 15 (2004).
[8] Lüdemann, A., D. Weicht, J. Selbig and J. Kopka: Pathway Visualization and Editing data base and software System. Bioinformatics Advance Access April 22 (2004).
[9] Thimm, O., O. Bläsing, Y. Gibon, A. Nagel, S. Meyer, P. Krüger, J. Selbig, L A. Müller, S. Y. Rhee and M. Stitt: MapMan: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. The Plant Journal 37, 914-939 (2004).
[10] Steinhauser, D. A. Lüdemann, J. Selbig and J. Kopka: Hypothesis-driven approach to predict transcriptional units from gene expression data. Bioinformatics Advance Access March 25 (2004).